Paving the way to better health and medical care
The Biophysics group from KTH Theoretical Physics has been using the GROMACS code to perform simulations of membrane proteins which may lead to more effective treatments for addiction disorders and safer anaesthetics.
by Erik Lindahl, Berk Hess, Mark Abraham, Szilard Pall and Magnus Andersson,
Science for Life Laboratory, Department of Biochemistry and Biophysics, Stockholm University,
Swedish e-Science Research Center, KTH Royal Institute of Technology
Erik Lindahl is the head of the Biophysics group in the Department of Theoretical Physics at the KTH Royal Institute of Technology and is also a professor of Biophysics in the Department of Biochemistry and Biophysics, Faculty of Science, at Stockholm University. Erik’s group is a large team whose research and development work spans the full gamut from fundamental methodology and software development for biophysics to the development of actual biological applications. The main people involved in the method and software development side of the research are Berk Hess (Associate professor), Mark Abraham (researcher), and Szilárd Páll (PhD student), all of whom are affiliated with the Theoretical Biophysics group within the Department of Theoretical Physics at KTH. On the application side, their software is used for large-scale simulations by a number of people, such as Magnus Andersson (researcher), who is also from Theoretical Biophysics. The whole team is closely affiliated with the Swedish e-Science Research Centre (SeRC) and the Science for Life Laboratory (SciLifeLab), both of which come under the Strategic research areas financed by the Swedish Research Council (VR) in the Stockholm region.

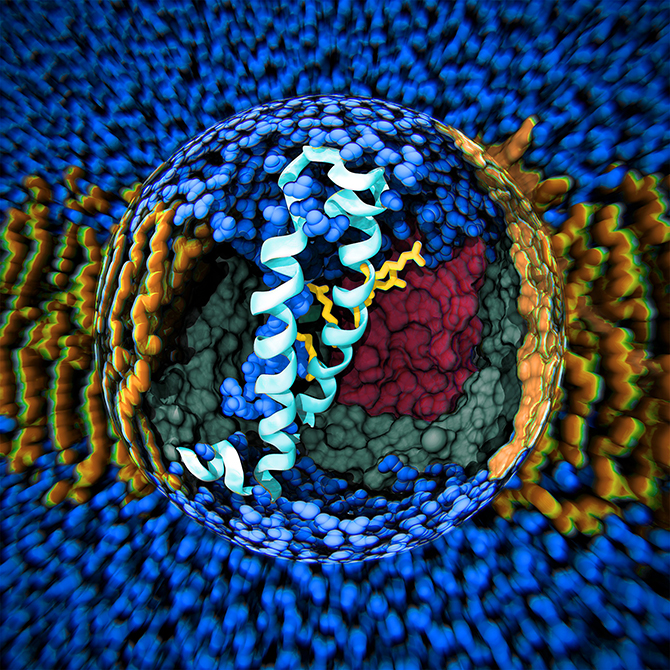
The team’s overall goal in advancing the state of the art in biomolecular modelling and simulation is to use these methods as a computational microscope for complex biological molecules. They are primarily working with membrane proteins – the small molecules that are embedded in our cellular walls and that act as the doors and windows of each cell. Every nerve signal and heartbeat in your body is due to these membrane proteins operating as microscopic machines and altering their conformations as a result of changes in the electric potential or ion concentration across the cell membrane. This behaviour is almost impossible to study experimentally since it only occurs transiently at the scale of microseconds. However, by using molecular simulations, it is possible to literally track the atomic-level motions to understand both how the proteins work and how we can change their behaviour.
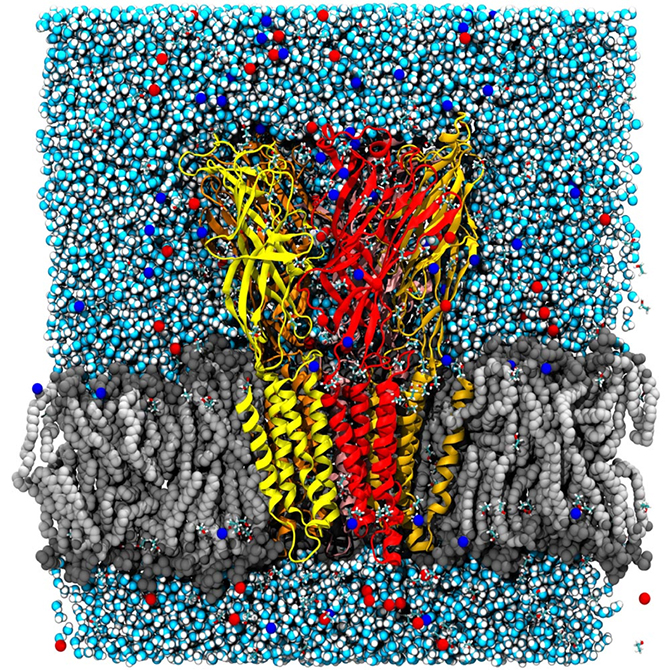
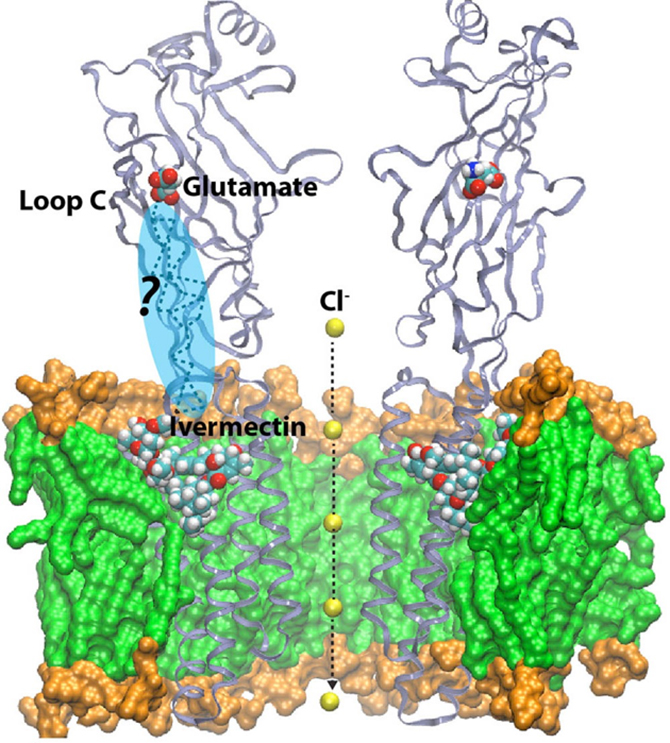
You might wonder what use that is in day-to-day terms! In actual fact, the entire early stage of the pharmaceutical drug design pipeline is carried out on computers these days, and GROMACS is used by thousands of academic researchers around the world, as well as by all the large pharmaceutical companies in the world. In just the last two to three years, Lindahl’s group has been able to use simulations to predict how the receptors in our nerve cells are regulated by external drugs such as anaesthetics and alcohol. This is a really complex process since some molecules up-regulate (or amplify) the response while others down-regulate (that is, dampen) the nerve signals. The drugs themselves have no effect whatsoever, but they act roughly like transistors: if they are present, the membrane protein channel will respond differently to the normal nerve signals. The research group were able to show that this odd-sounding effect is due to multiple different binding sites in the membrane part of the protein, which have opposite effects. This might sound technical and advanced, but the cool result is that it may make it possible to design new pairs of drugs which would, for example, enable us to fine-tune anaesthesia to safely sedate elderly or ill patients (where anaesthetics can sometimes have unexpected effects compared to with patients in a normal state of health). This discovery also has the potential to improve the treatment of many addiction disorders, including alcoholism as ethanol hits these ligand-gated channels!