Materials Theory Codes on Dardel at PDC
Johan Hellsvik, PDC, Olle Eriksson, Department of Physics and Astronomy, Uppsala University, & Igor Di Marco, Asia Pacific Center for Theoretical Physics
Quantum mechanical modelling based on density functional theory (DFT) has evolved to be a cornerstone of computational materials science, which is applicable to a wide range of functional materials, quantum materials, and materials relevant to green technologies. DFT provides a tractable means to model the complex interactions of a quantum mechanical many-body system.
Within a pre-production project that has been running on the new Dardel HPE Cray EX supercomputer at PDC (with Olle Eriksson as the principal investigator), codes developed at the Materials Theory Division at Uppsala University have been installed and tested on the CPU-partition of Dardel. The codes are being developed in close collaboration with groups at Örebro University, the KTH Royal Institute of Technology, the Federal University of Pará in Belém, Brazil, the University of Sao Paulo, Brazil, and the Asia Pacific Center for Theoretical Physics (APCTP), South Korea. These codes include the full-potential all-electron DFT programs RSPt and Elk, and the atomistic spin-dynamics program UppASD. These codes also involve the augmentation of electronic structure theory to include effects of many-body physics – in the form of dynamical mean field theory – which is an effort that involves in-house and externally developed quantum many-body solvers.
Whereas the atomistic spin-dynamics approach – which implements a lattice model for particles (spins) interacting with short-range interactions – is naturally suited for partitioning the problem volume over distributed computer memory (with parallel sampling and time evolution of the system on the nodes of a supercomputer), the DFT programs implement models with interactions on both shorter and longer length scales. With regard to the effective scaling of program performance towards exascale computing, the DFT programs and algorithms therefore pose a challenge, as performance is dependent on – and in part limited by – the need for individual execution threads and ranks of a program to access state variables that are global to the physical model.
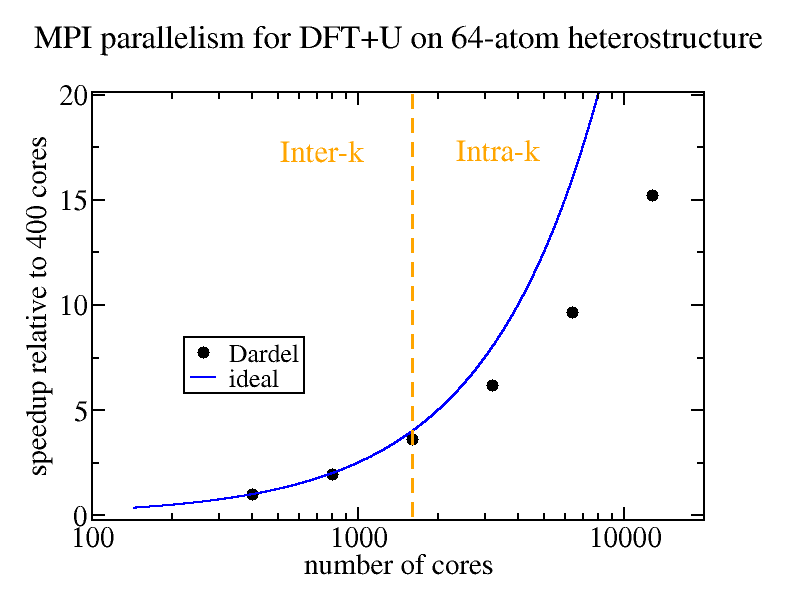
The focus for the present project is to tune the performance on the Dardel CPU nodes, while keeping an eye out for chances to explore possible opportunities to adopt kernels of the materials theory codes to the Dardel GPU nodes in the future. Solving the Schrödinger equation in reciprocal space, a DFT program can be parallelized over a finite set of wave vectors (k points) within the Brillouin zone of the material. As an example from using the Dardel CPU nodes, for the full-potential linearized muffin-tins orbitals code RSPt, the strong scaling performance for algorithmically and memory demanding intra k-point parallelization – as well as the more tractable inter k-point parallelization – are under investigation, with preliminary findings indicating good performance as well as opportunities for further improvement. Nevertheless, the tests have also shown that the optimal number of processes per node for maximizing performance is half the number of available cores, which points to some delay in the MPI communication. Work to minimize these delays will proceed by exploring additional combinations of compilers and libraries, and further tuning of the code.