VeloxChem: Efficient Numerical Integration for Density Functional Theory
Xin Li, PDC, and Zilvinas Rinkevicius & Patrick Norman, Division of Theoretical Chemistry and Biology, KTH
Density functional theory (DFT) is a widely used theoretical framework in the field of computational chemistry due to its versatility in the study of various systems and properties. It offers a favourable trade-off between accuracy and computational efficiency, which makes it particularly useful for investigating large and complex systems. DFT has, therefore, become an indispensable tool in scientific research and theoretical design of molecules and materials.
VeloxChem [1] is an open-source quantum chemistry software that was developed at the KTH Royal Institute of Technology for calculating molecular properties and simulating a variety of spectroscopies. VeloxChem offers DFT as one of its key features. In recent developmental work on the code, we have improved the efficiency of DFT numerical integration within VeloxChem, so now the exchange-correlation part can be run on computer systems featuring either CPUs or graphics processing units (GPUs). Conventionally, the computational cost of DFT is proportional to N3 where N is the size of the system being studied; however, the numerical grid-based integration can, in practice, be done very efficiently by integrating over even-sized grid batches, such that the computational cost becomes asymptotically linear [2]. The key to such an efficient implementation is to make sure that the whole system is divided as evenly as possible into grid batches with almost constant computational cost on average. In VeloxChem this is done by repeated bisection of grid points into smaller and smaller boxes until the number of grid points in each box is below a given threshold. Due to efficient screening, the computational cost for processing each grid box will be almost constant on average, making it highly suitable for large-scale parallelisation.
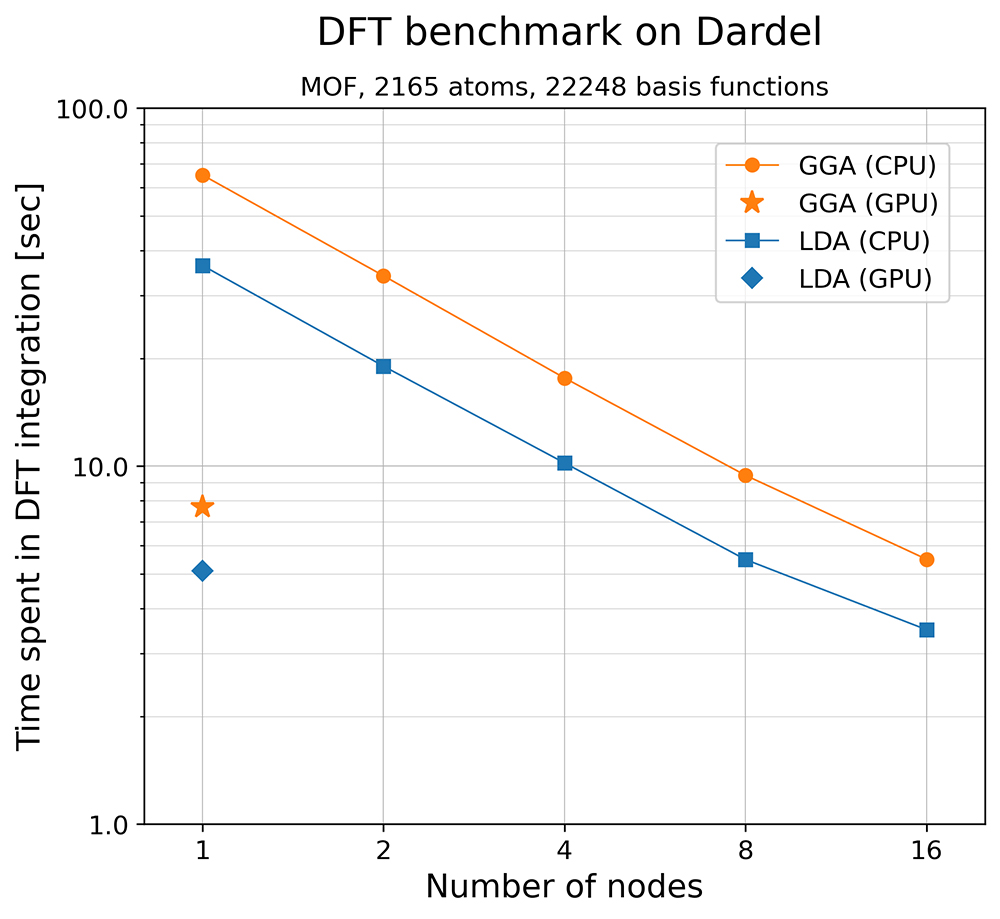
We tested the performance of DFT numerical integration in VeloxChem using a metal-organic framework (MOF) structure (illustrated above) that consists of more than 2,000 atoms. This is a very large system for quantum chemistry, and the number of basis functions exceeds 22,000, even for a moderate-sized basis set. Two flavours of DFT numerical integrations were tested on the Dardel compute nodes: the local density approximation (LDA), which is a rather crude approximation, and the generalised gradient approximation (GGA), which is more widely used in practice. Both LDA and GGA were tested by varying the number of Dardel CPU nodes that were used, and one can see from the solid lines in the graph below that the computational time decreases almost linearly in the logarithmic scale plot. We also tested the performance of LDA and GGA on a Dardel GPU node, as shown by the star and diamond markers on the graph. On a single GPU node, the time spent in LDA and GGA integration are 5.2 and 7.7 seconds, respectively. This is significant as it opens up the possibility of routine studies of large and complex chemical systems for scientific research in biochemistry, nanoscience and spectroscopy.
References
- Z. Rinkevicius, X. Li, O. Vahtras, K. Ahmadzadeh, M.Brand, M.Ringholm, N.H.List, M.Scheurer, M. Scott, A. Dreuw, P. Norman. “VeloxChem: a Python-driven density-functional theory program for spectroscopy simulations in high-performance computing environments”. WIREs Comput. Mol. Sci. 2020, 10:e1457. doi.org/10.1002/wcms.1457
- J. Kussmann, H. Laqua, C. Ochsenfeld. “Highly Efficient Resolution-of-Identity Density Functional Theory Calculations on Central and Graphics Processing Units”. J. Chem. Theory Comput. 2021, 17, 3, 1512–1521. doi.org/10.1021/acs.jctc.0c01252