VeloxChem — Quantum Molecular Modelling in HPC Environments
Zilvinas Rinkevicius, Xin Li, and Patrick Norman, Department of Theoretical Chemistry and Biology, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH
Vision and Goals
Theoretical chemistry has made tremendous progress in the past few decades and is today an indispensable tool in all fields of molecular science, exemplified by biochemistry and nanotechnology, where it can be employed to reveal the microscopic origins of functionality and interactions as well as to tune performance and guide synthesis. The interplay between simulation and experiment is a key enabling factor for advancements in the life and materials sciences, and spectroscopy represents one of the most important meeting points in between the two disciplines.1 It is imperative to base spectroscopic modelling on first principles, as the underlying microscopic events are quantum-mechanical in origin2— although typically in combination with classical force field molecular dynamics for the sampling of configuration space. In order to study complex molecular systems and delocalised electronic transitions, it is consequently important that such first-principles approaches are developed into forms where they may be tractably applied to large systems. It is therefore not surprising that a vast amount of work in computational chemistry has been carried out to extend the computationally tractable system domain boundaries with respect to time and length scales.3
Alongside the development of methods and algorithms, we are witnessing an ever ongoing advancement of high-performance computing (HPC) cluster resources to reach higher numbers of floating point operations per second. In the first PDC Newsletter for 2019, PDC Director Erwin Laure presented the EuroHPC project and its leading Europe toward exascale computing ( www.pdc.kth.se/publications/pdc-newsletter-articles/2019-no-1/eurohpc-europe-s-path-to-exascale-1.911735 ). EuroHPC comprises an entire supercomputing ecosystem with the ambition to strengthen European industry and academia alike, and as such it brings us an exciting future at the same time as it challenges us. A view of the most recent TOP500 list gives a clear indication that heterogeneous hardware solutions are to be expected when such extreme performance numbers are to be reached and there is a community consensus among quantum chemists that an efficient utilization of general-purpose graphical processing units (GP-GPUs) is difficult to achieve in our software where the compute-intensive kernels involve 104–105 lines of code. What we believe to be the competitive edge of the VeloxChem project in this respect is the fact that it started from scratch some two years ago without the burden of a code legacy.
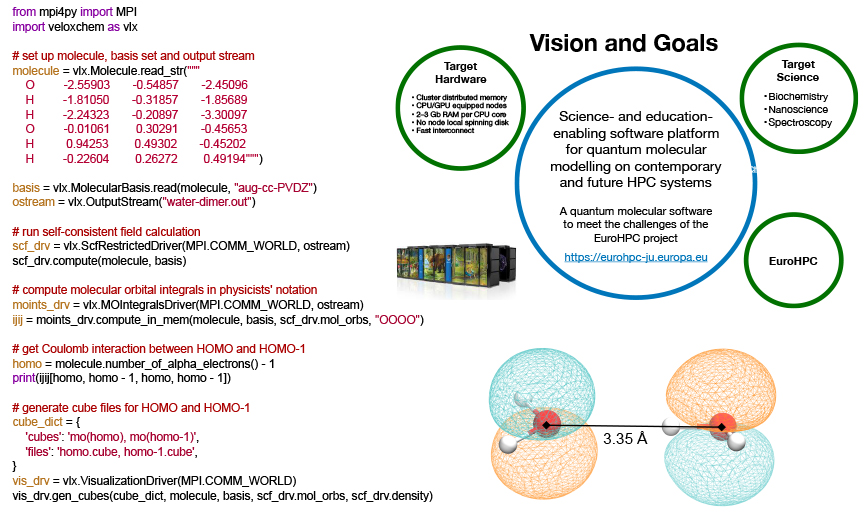
The stated goals of the VeloxChem program (which are displayed in the figure above) encompass the following:
Behind the term science-enabling there are a multitude of software requirements that we find important in our work, including (i) coverage of system sizes up to and beyond 500 atoms in the quantum region, (ii) accurate description of electronically excited states that show a more diffuse character than the ground state, (iii) stable and reliable convergence of iterative equation solvers also with use of diffuse basis sets, (iv) time-efficient prototyping of novel scientific approaches, (v) transparent exposure of data structures to enable in-depth analyses for standard users, (vi) flexible ways to interact with other components of the simulation (such as molecular dynamics, parameterizing the embedding, and data visualization) and, not least, (vii) a fast return of results so as to remain in synchronicity with experimental project partners. The term education-enabling adds another set of software requirements to this already long list. In this context, the notion of deeper learning refers to taking each student’s understanding of the subject matter to another (deeper) level. Our experience tells us that the process of implementing methods to solve fundamental equations is supremely efficient as a means to achieve that type of deeper learning, but only a small number of students are granted this opportunity as many core modules of scientific software were written a long time ago and have often been made obscure by code optimization. What if we could instead offer access to the needed building blocks to explore quantum chemistry in very much the same manner that we can use the Python NumPy package to explore linear algebra?
All of these considerations went into the design of the VeloxChem program. We started out with the ambition to (i) scale standard spectroscopy calculations beyond 10,000 central processing unit (CPU) cores so as to be considered relevant for the EuroHPC project and, at the same time, (ii) provide a platform for undergraduate students to implement (in Python, on a laptop) their own self-consistent field (SCF) optimizer of ground state wave functions within the limited time of a course exercise. We have now reached these goals and a more detailed presentation of the resulting VeloxChem program has been accepted for publication in the Wiley Interdisciplinary Reviews (WIREs).4
Program Structure and Modularity
From a programming language perspective, the VeloxChem program is designed in a two-layered fashion. The upper layer is written in Python and manages the use of the available hardware resources by means of dynamically split message passing interface (MPI) communicators for separate and parallel execution of the three compute-intensive components of the calculation namely (i) the analytical evaluation of electron-repulsion integrals (ERIs), (ii) the numerical grid-based integration of the Kohn–Sham density functional theory (DFT) exchange-correlation kernel, and (iii) the contribution due to the coupling between the quantum mechanical (QM) focus region of the system and the classical molecular mechanics (MM) embedding region (described by point charges and atomic-site polarizabilities). Any one of these three components will scale with the size of the hardware resource only up to a point and, by using split communicators and parallel execution, we can scale the calculation further and reach shorter wall times of execution. The splitting of the communicator is best made dynamic because every model system — depending on the relative sizes of the QM and MM regions, basis set, grid accuracy, and so forth — will experience different and hardware-dependent (CPU versus GPU) load balancing that needs to be adjusted for in order to avoid idling resources.
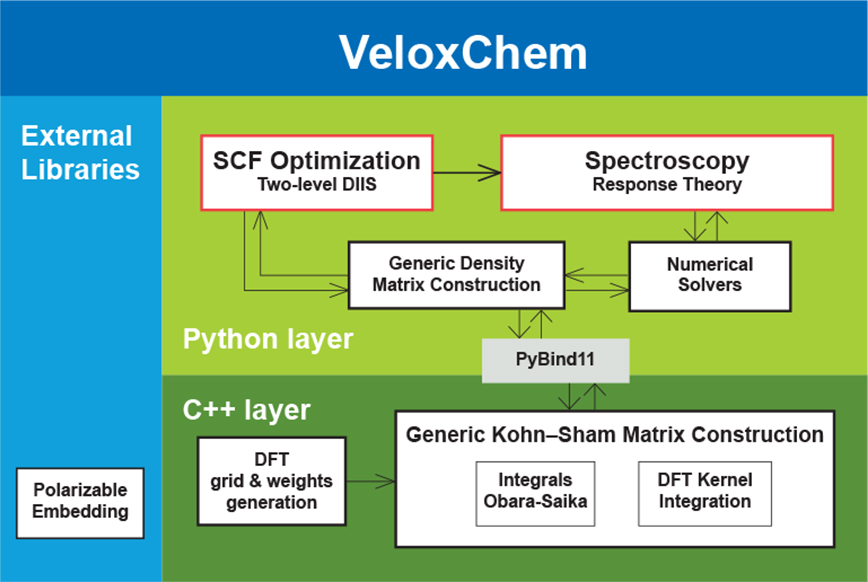
The Python layer also implements high-level quantum chemical methods such as (i) the direct inversion in the iterative subspace (DIIS) technique to reach the self-consistent field (SCF) solution of the ground-state electron density and (ii) the several variants of iterative numerical response equation solvers that provide the spectral responses (see the figure above). Communication of data arrays between the Python and C++ layers is achieved with the header-only library Pybind11.5 All underlying algorithms (SCF and response) are formulated such that there is one common set of data arrays that needs to be communicated through the layers, namely generalized density matrices from Python to C++ and the corresponding Fock matrices (after a contraction is made with the ERIs) from C++ to Python. However, this design also brings a more novel feature to the VeloxChem program as we may expose virtually any data array from the C++ layer to be accessed by the user as a NumPy6 array in the Python layer. As a result, not only are students equipped with the tools to perform educational exploratory work as discussed in the introduction but general users can also perform in-depth analyses of their research. As an illustration of the latter aspect, the first figure in this article shows how it is possible, with a Python code snippet, to set up the calculation of a water dimer system and obtain the Coulomb interaction energy between the electrons occupying the highest occupied molecular orbitals (HOMOs) of the respective water molecules. For the water dimer configuration under consideration, this interaction energy amounts to the following ERI:
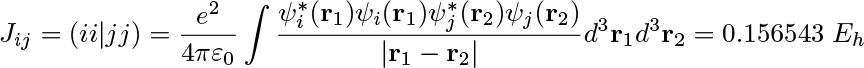
which, incidentally, is rather close to the interaction energy of 0.158 Eh for two elementary point charges separated by the O–O distance of 3.35 Å. We believe that every experienced computational scientist working with monolithic legacy codes can appreciate the ease with which the VeloxChem program provides transparent access to specific user-requested intermediate data in the calculation, extracted from arrays that, due to the sheer number of elements, would never be stored or made accessible to the user. At this point, the question to be asked is whether this code design comes at the price of reduced computational efficiency and we will address this question in the next sections.
Vectorization and Parallelization
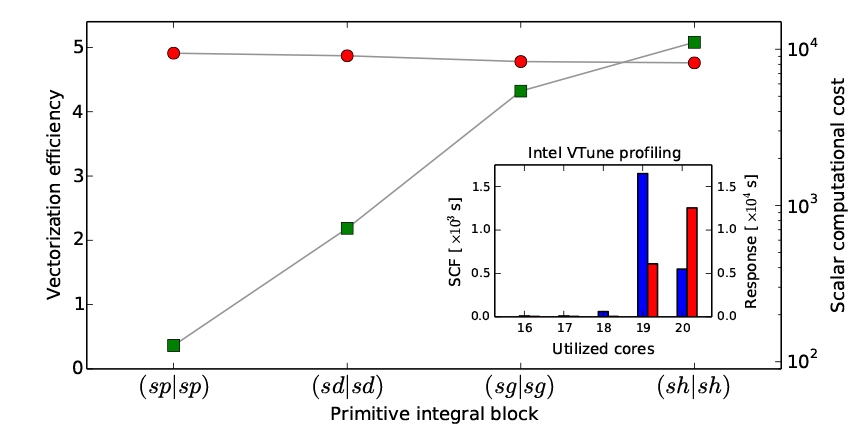
VeloxChem employs a modified version of the Obara–Saika scheme7 to analytically evaluate ERIs. The implementation is based on automated code generation with a high degree of loop structure optimization in order to achieve efficient vectorization on CPUs that implement single instruction multiple data (SIMD) instructions. The obvious goal of this vectorization is to achieve efficient usage of the vector registers on modern CPUs. In the figure below, we present a performance profile of the vectorization for the vertical recursion step in the workflow scheme. For basis set primitives up to angular momentum l = 5 (or h-functions), the efficiencies reported by the Intel-compiler and depicted in the figure exceed a speedup of 4.75 for the vectorized code as compared to the scalar code.
Vectorization Efficiency
The OpenMP parallelization on a single node is benchmarked using the Intel VTune Amplifier tool that measures the amount of time spent at different levels of CPU core activity. Thus, the range of activity spans from zero (fully idling CPUs) to the maximum number of cores on the node (in our case 20). The inset in the figure below depicts the reported results for the complete SCF optimization of the Hartree–Fock ground state of the fullerene C60, as well as the subsequent complete response calculation to obtain the five lowest roots of the generalized eigenvalue equation, that is, the five lowest singlet states in the UV/vis spectrum. The overall total OpenMP threading efficiency is seen to reach levels of some 96% and 98% for the SCF (blue bars) and response parts (red bars), respectively.
In the next figure, we address a titanium oxide nanoparticle that illustrates the size (but not necessarily type) of system that we routinely wish to encompass in the quantum region of a given physical model. With generous and exclusive access to a large part of the main cluster at the PDC Center for High Performance Computing in Stockholm, we had the opportunity to test the MPI/OpenMP scaling of VeloxChem up to 16,384 cores (or 512 dual socket 32-core Intel Xeon nodes) and we used the otherwise identical calculation performed on 32 cores (or 1 node) as the benchmark reference. A complete SCF cycle during the optimization of the ground state density requires a wall time of 70,565 s in the reference calculation, out of which 70,493 s and 72 s are spent on the construction of the Fock matrix and other parts (including the Fock matrix diagonalization) respectively. Apart from the Fock matrix construction, the SCF module is written in Python (as shown in the VeloxChem program structure diagram earlier) and executes on a single node and the corresponding timing is therefore nearly constant along the series of calculations presented in the figure below.
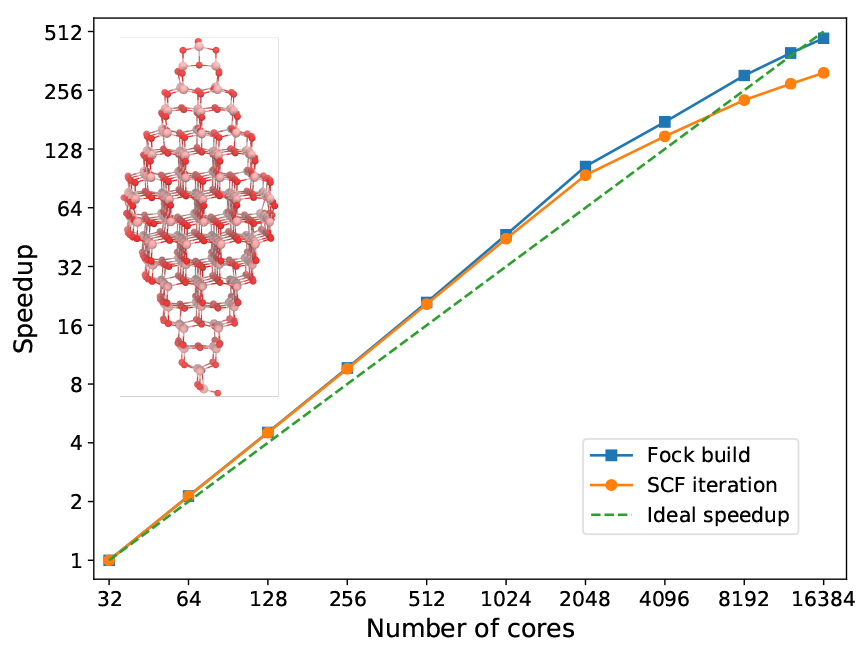
Owing to favourable differences in the dynamic workload balancing between the available MPI processes and OpenMP threads, the Fock matrix construction scales slightly beyond linear with respect to the number of cores (up to a point of 384 nodes) and reaches an efficiency of 162.9% at the point of using 64 nodes and compared to the reference calculation. This example thus demonstrates almost perfect scaling up to the point of allocating almost one CPU core per primitive basis function. The wall time for the Fock matrix construction using 512 nodes is reduced to 149 s, so while it may be possible to observe a reasonable scaling beyond 512 nodes in the present example; it makes little sense in practice as the SCF/DIIS part becomes comparable in duration.
Spectrum Calculations
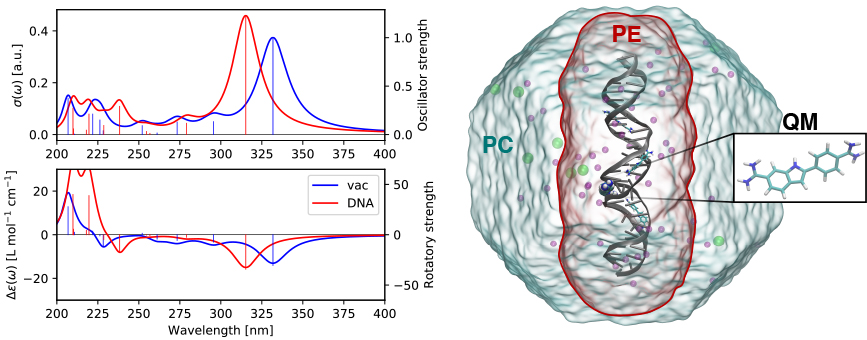
As a single example of the scientific capabilities of VeloxChem, the figure below shows the calculated UV/vis and electronic circular dichroism (ECD) spectra of the DAPI biomarker inside the minor groove of DNA as studied in an earlier paper.8 The model system illustrated in the figure is set up with (i) a DAPI biomarker in the QM region, (ii) a polarizable embedding (PE) region containing the DNA sequence, the three additional bound DAPIs, and a 15 Å shell of water molecules with a small number of sodium and chlorine ions (the entire system is charge neutral), and (iii) the remaining water molecules and ions in the surrounding point-charge (PC) region. In total, the model includes some 85,000 and 23,000 point-charge and polarizable sites, respectively.
Outlook
With the publication of the program release paper4, VeloxChem version 1.0 will be distributed under the GNU Lesser General Public License version 2.1 (LGPLv2.1) licence and made available for download from the VeloxChem home page veloxchem.org . At present, VeloxChem offers spectrum simulations of complex molecular systems on CPU-based HPC cluster facilities. With a layered Python/C++ language design, it achieves excellent MPI/OpenMP parallel scaling and SIMD vectorization for realistic systems and using up to a quarter of the Beskow cluster. The compute- intensive integral code is large but remains agile and adaptable to new hardware solutions owing to the fact that it is based on automated code generation.
A main future focus point is to develop CUDA implementations of the three compute-intensive components of VeloxChem namely (i) analytic evaluation of integrals, (ii) numerical DFT kernel integration, and (iii) polarizable embedding. Out of these three, the implementation of integrals is design-wise the difficult one. We are grateful for the kind invitation to participate at the Eurohack19 workshop in Lugano (which ran from the 30th of September to the 4th of October 2019), where we received general guidance and dedicated expert help to start on this endeavour. During this intense week, we implemented a first version of a code for integral evaluation in the primitive Gaussian orbital basis with Cauchy–Schwarz screening, and we saw tentative performance numbers that point toward a great future for VeloxChem in the landscape of heterogeneous cluster computing.
Acknowledgements
The VeloxChem project is enabled by our participation in the Swedish e-Science Research Centre (SeRC) and by integrated collaboration with the PDC Centre for High Performance Computing. The results presented in this article were obtained with the use of computational resources provided by the Swedish National Infrastructure for Computing (SNIC).
References
- Lindon, J. C., Tranter, G. E., Holmes, J. L., Eds. Encyclopedia of spectroscopy and spectrometry; Academic Press: San Diego, 2010.
- Norman, P.; Ruud, K.; Saue, T. Principles and practices of molecular properties; John Wiley & Sons, Ltd. Chichester, UK, 2018.
- Akimov, A. V.; Prezhdo, O. V. Large-Scale Computations in Chemistry: A Bird’s Eye View of a Vibrant Field. Chem. Rev. 2015, 115, 5797–5890.
- Rinkevicius, Z.; Li, X.; Vahtras, O.; Ahmadzadeh, K.; Brand, M.; Ringholm, M.; List, N. H.; Scheurer, M.; Scott, M.; Dreuw, A.; Norman, P. VeloxChem: a Python-driven density-functional theory program for spectroscopy simulations in high-performance computing environments. WIREs Comput. Mol. Sci. 2019, DOI: 10.1002/wcms.1457.
- Jakob, W.; Rhinelander, J.; Moldovan, D. pybind11 – Seamless operability between C++11 and Python. 2017; github.com/pybind/pybind11 .
- van der Walt, S.; Chris Colbert, S.; Varoquaux, G. The NumPy array: A structure for efficient numerical computation. Comput. Sci. Eng. 2011, 13, 22–30.
- Obara, S.; Saika, A. Efficient recursive computation of molecular integrals over Cartesian Gaussian functions. J. Chem. Phys. 1986, 84, 3963–3974.
- List, N. H.; Knoops, J.; Rubio-Magnieto, J.; Idé, J.; Beljonne, D.; Norman, P.; Surin, M.; Linares, M. Origin of DNA-Induced Circular Dichroism in a Minor-Groove Binder. J. Am. Chem. Soc. 2017, 139, 14947–14953.